Medical Sciences
Decoding Waardenburg Syndrome,Genetic Aspects and Clinical Nuances of Waardenburg Syndrome

Understanding Waardenburg Syndrome: Genetic Aspects and Clinical Insights
Introduction
Waardenburg syndrome stands as a rare genetic disorder renowned for its distinctive features involving sensorineural hearing loss and pigmentary abnormalities affecting the hair, skin, and eyes. Named after the Dutch ophthalmologist Petrus Johannes Waardenburg, who first observed the correlation between deafness and heterochromia iridis, this syndrome has been a subject of keen medical interest.
RESOURCED ARTICLE Waardenburg syndrome

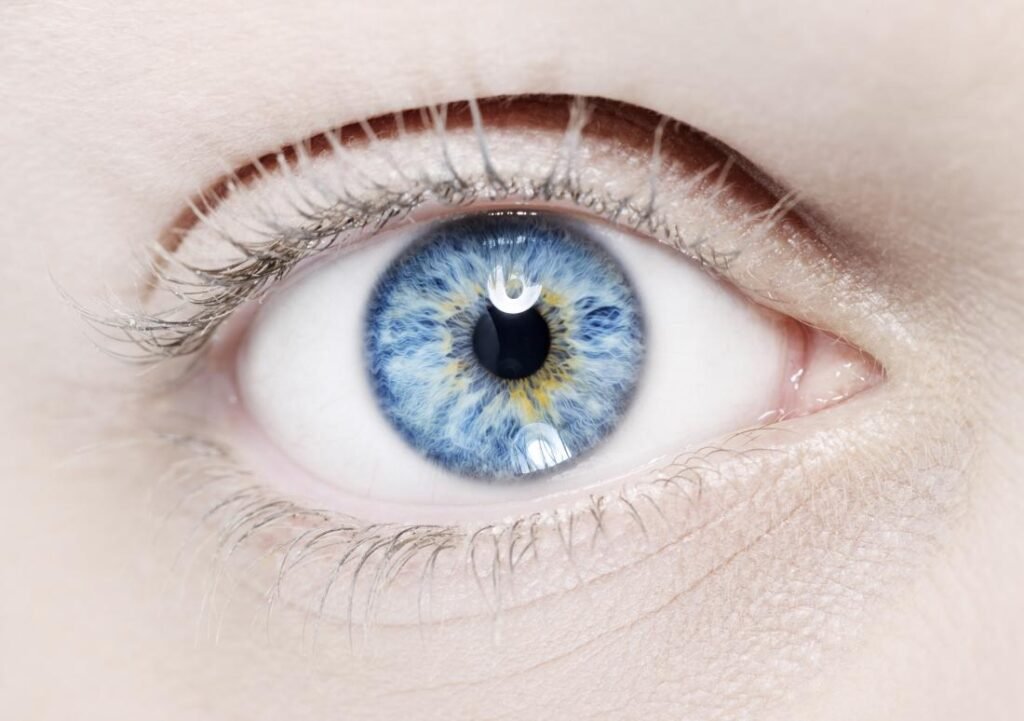
Demographics and Frequency: Waardenburg syndrome does not discriminate; it affects both males and females equally and can manifest across all races. With a population frequency of approximately 1 in 40,000, it contributes to 2–5% of all cases of congenital deafness.
Genetic Basis: This neurocristopathy arises from gene mutations affecting neural crest differentiation during embryonic development. Various genes play pivotal roles, and their mutations give rise to different types of Waardenburg syndrome. Noteworthy genes include:
- PAX3 (Chromosome 2q): Implicated in WS1 and WS3.
- MITF (Chromosome 3p): Associated with WS2A.
- Chromosomes 1p, 8p, 8q, 13q, 20q, and 22q: Linked to WS2B, WS2C, WS2D, WS4A, WS4B, and WS4C, respectively.
The mutations encompass various types such as insertions, deletions, frameshifts, splice alterations, missense, or nonsense mutations. Interestingly, most types of Waardenburg syndrome follow an autosomal dominant inheritance pattern, meaning that one affected gene passed on to a child is sufficient for them to exhibit the syndrome. However, complexities arise with genes like EDN3 or EDNRB, which generally follow autosomal recessive patterns.

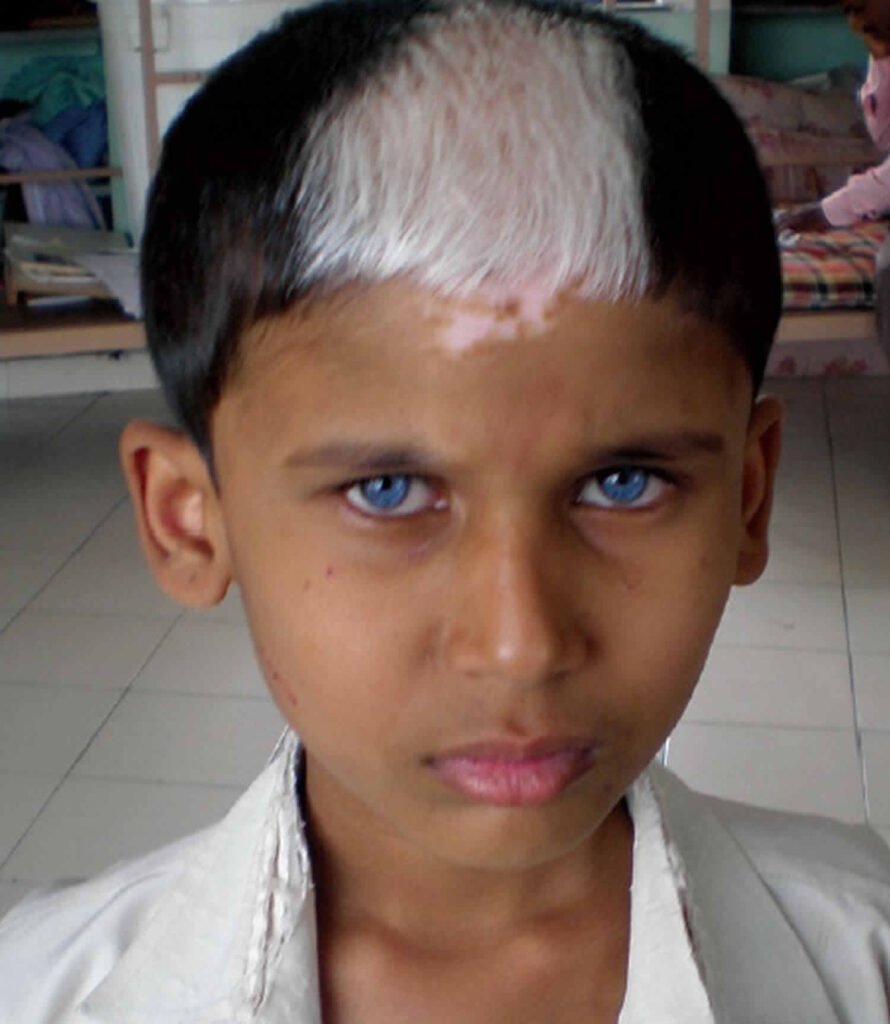
Clinical Features: Waardenburg syndrome manifests early in life, and its diagnosis can be challenging due to the subtlety of clinical signs. Major types (Type 1-4) display distinct features:
- Type 1: Most common, characterized by sensorineural deafness, distinct facial features, white skin patches, and pigmentary abnormalities in the eyes.
- Type 2: Similar to Type 1 but lacks abnormalities in the inner canthi.
- Type 3 (Klein-Waardenburg syndrome): Includes musculoskeletal abnormalities.
- Type 4 (Shah-Waardenburg syndrome): Similar to Type 2 but with the addition of Hirschsprung syndrome.
Diagnosis: The diagnosis of Waardenburg syndrome relies on clinical features, with major criteria encompassing sensorineural deafness, iris pigmentary abnormalities, and hair pigmentation anomalies. Minor criteria include features like unpigmented skin patches, synophrys, and premature greying of scalp hair. The W index, calculated based on canthal distances, aids in identifying dystopia canthorum. Genetic sequencing, particularly of the PAX3 gene, can provide a definitive diagnosis and guide genetic counseling.


Treatment and Management: As of now, there is no curative treatment for Waardenburg syndrome due to its genetic nature. Genetic counseling is crucial for affected individuals contemplating starting a family. Regular audiology examinations are recommended for managing deafness, and those with Hirschsprung syndrome may require surgery. Sun protection is emphasized for areas with unpigmented skin patches.
Outlook: Waardenburg syndrome presents stable clinical features throughout life, and individuals can expect a normal life expectancy. Although the syndrome poses challenges, advancements in genetic understanding have paved the way for improved diagnostics and counseling, offering individuals and families affected by Waardenburg syndrome a clearer path forward.
READ MORE INTERESTING ARTICLE Cortex Chronicles, Paul G. Allen’s Institute Explores the Study on Visual Masking in Bioscience
In conclusion, the intricate genetic landscape of Waardenburg syndrome underscores the importance of ongoing research and medical advancements in unraveling the complexities of this rare disorder. As we delve deeper into the genetic intricacies, our ability to diagnose, manage, and provide support for individuals with Waardenburg syndrome continues to evolve.